
Exploring the chemical space
The chemical space of drug-like molecules is a mind-boggling >10⁶⁰ compounds [1] — a cosmic expanse where traditional methods falter. Yet, how much of this is accessible through the Anyo Lab’s molecular generator (MolGen)? Here we utilize mathematical frameworks, some borrowed from ecology, to estimate the number of unique chemically valid molecules it can generate.
A Blend of Ecology and Computation
To estimate the total number of unique molecules from a large sample is an analogous task to estimating the number of species from a set of observations, where each unique molecule is counted as a species. To empirically estimate the chemical space of MolGen, we generated 1 billion chemically valid molecules and utilized three species-estimators: Chao1 [2], ACE [3], and Good-Turing [4].
When the estimators’ predictions were analyzed, it showed that they continuously update their predictions to larger numbers with increasing sample sizes, indicating that 1 billion molecules is insufficient for convergence. The final estimations from Chao1, ACE, and Good-Turing were: 1 × 10¹⁰, 7.9 × 10⁹, and 2.5 × 10⁹ unique molecules. It became clear that when a large fraction of the molecules are unique (75.3% for 1 billion molecules), the estimators underestimate the true explorable chemical space.
To overcome the species-estimators' limitations, a second method models the unique fraction of molecules as a logarithmic function of the number of generated molecules (which appears linear on a logarithmic x-axis) and it is extrapolated as far as reasonable, as shown in Figure 1.
The product of the extrapolated unique fraction and the generated number yields the unique number of molecules, which estimates a lower bound prediction of 1 × 10¹⁴ molecules. This model represents a fair trade-off between simplicity and accuracy.
However, an obvious limitation of this model is that it underestimates the true unique fraction of molecules, as it will extrapolate to impossible values such as zero uniqueness near 10¹⁶ generated molecules, whereas the true function for the unique fraction should resemble an S-curve (as observed for the unique scaffolds). This cements 10¹⁴ as the lower bound.
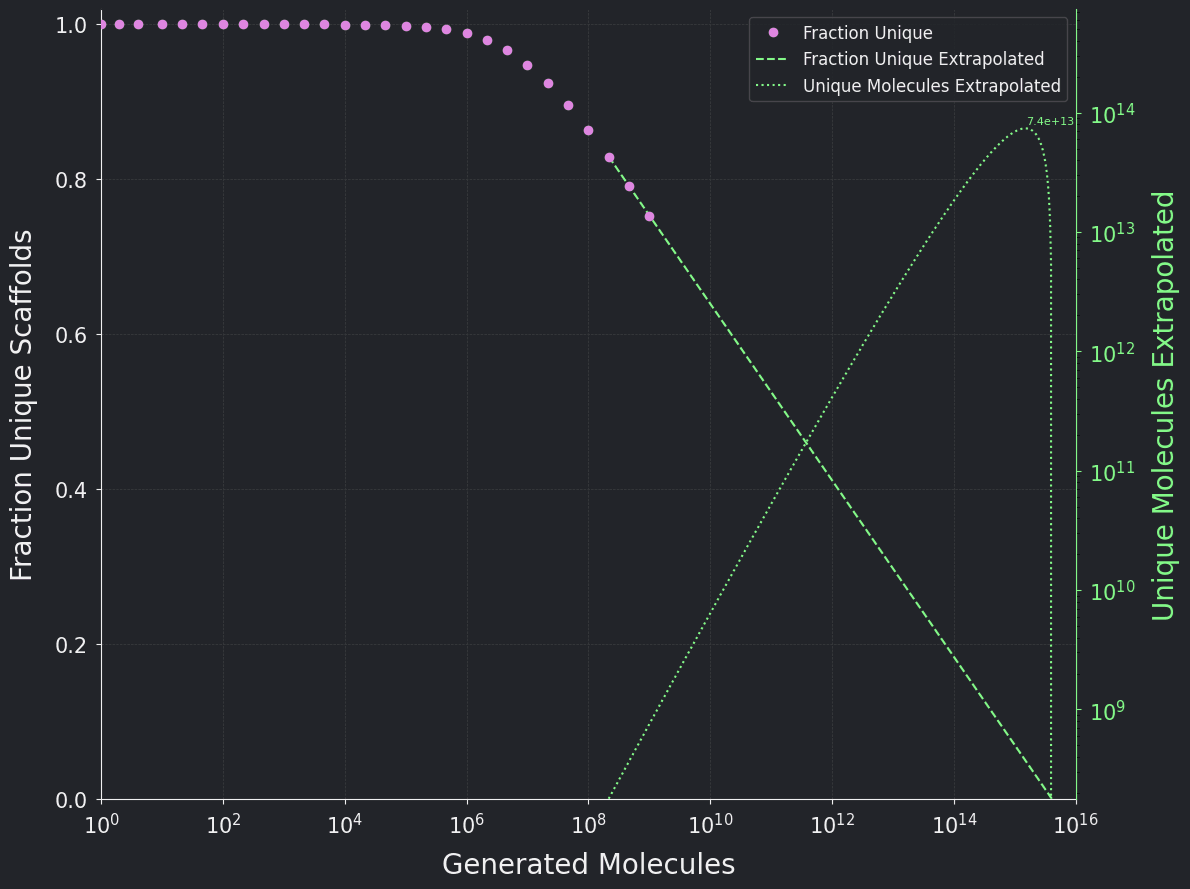 Figure 1. The fraction of unique molecules is displayed in pink over the number of generated molecules in log-scale. A simple extrapolation of the unique fraction of molecules and the unique number molecules are displayed in green.
Figure 1. The fraction of unique molecules is displayed in pink over the number of generated molecules in log-scale. A simple extrapolation of the unique fraction of molecules and the unique number molecules are displayed in green.
Estimate Number of Unique Scaffolds and Molecules from Direct Extrapolations
To analyze the diversity, we computed the unique scaffolds of the 1 billion generated molecules and applied both methodologies described above. Three types of scaffolding techniques were utilized:
- True Murcko Scaffolds, which only extract the ring systems and their linkers without any double-bonded atoms.
- RDKit Murcko Scaffolds, which are True Murcko Scaffolds that include double-bonded atoms (to guarantee chemical validity).
- Generic Scaffolds, which are True Murcko Scaffolds where all atoms and bonds are carbon and single bonds.
Just as the species-estimators did not converge on the estimated number of molecules, none of them converge on the number of scaffolds either. Using the same methodology to extrapolate the unique fraction, the predicted minimum number of unique scaffolds for RDKit, True, and Generic scaffolds were calculated to be:
- 1.1 × 10¹⁰ scaffolds (RDKit)
- 6.5 × 10⁹ scaffolds (True Murcko)
- 1.2 × 10⁸ scaffolds (Generic)
Furthermore, we do not observe a large decline in the number of generated unique scaffolds for increasing sample sizes, as shown in Figure 2. This indicates that MolGen can generate orders of magnitude more unique scaffolds than were sampled in this study.
Figure 2. Number of unique scaffolds over number of generated molecules in log-log scale. Generic, True Murcko, or RDKit Murcko scaffolds show a very slight decline for increasing numbers of generated molecules.
As Figure 2 indicates, the datapoints do not follow a completely straight line. One should expect the data series to flatten out for large magnitudes of generated molecules when it approaches the size of the explorable chemical space. Fitting a “quadratic-exponential” function:
𝛼 ⋅ (10ˣ)² + 𝛽 ⋅ (10ˣ)
to the scaffold data points, we extrapolate to get an estimated size of the scaffold-chemical space for each scaffold type, as shown in Figure 3.
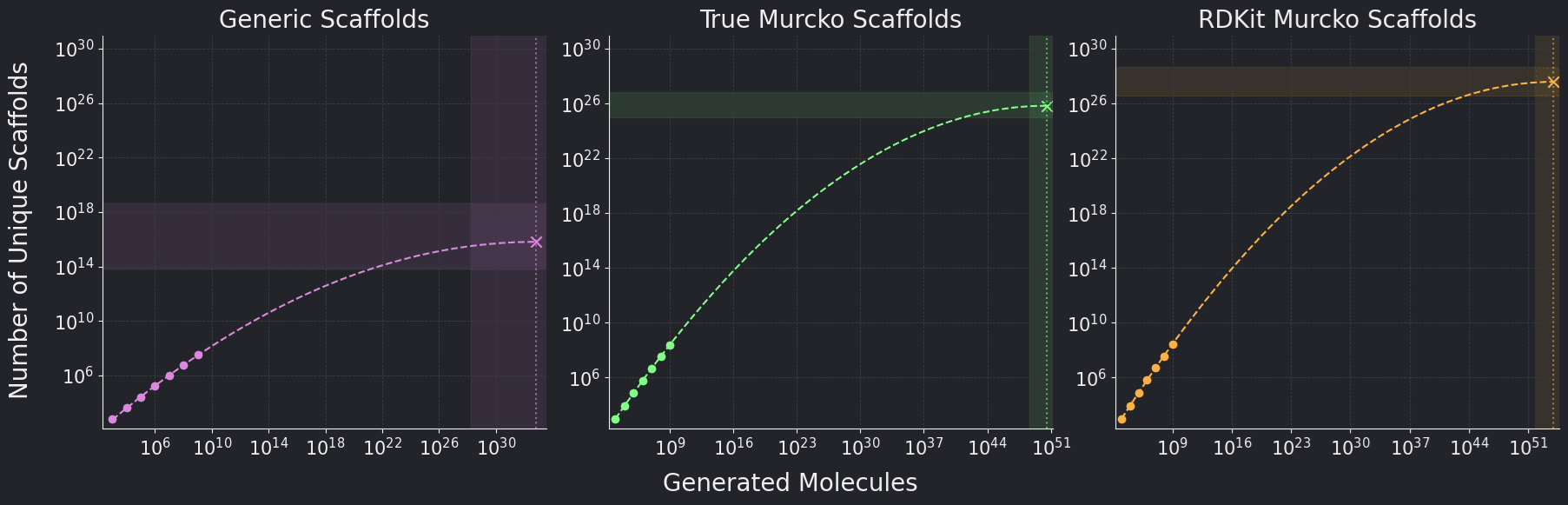 Figure 3. Extrapolated number of Generic, True Murcko, and RDKit Murcko Scaffolds over
number of generated molecules. The 95% confidence interval of the maximum value of the
fit is displayed in each plot.
Figure 3. Extrapolated number of Generic, True Murcko, and RDKit Murcko Scaffolds over
number of generated molecules. The 95% confidence interval of the maximum value of the
fit is displayed in each plot.
Fitting the same model to the number of unique molecules and generated molecules gave an unreasonably large estimation of MolGen’s chemical space because the data points were very close to linear making it difficult to accurately fit the curvature-constant . However, by modelling the number of unique molecules per unique scaffold over the generated number of molecules (with a linear equation) and multiplying this with the extrapolated maximum number of scaffolds, it yields an estimation for number of unique molecules for each scaffold.
Table 1. Estimated number of unique molecules from extrapolated number of scaffolds and molecules per scaffold.
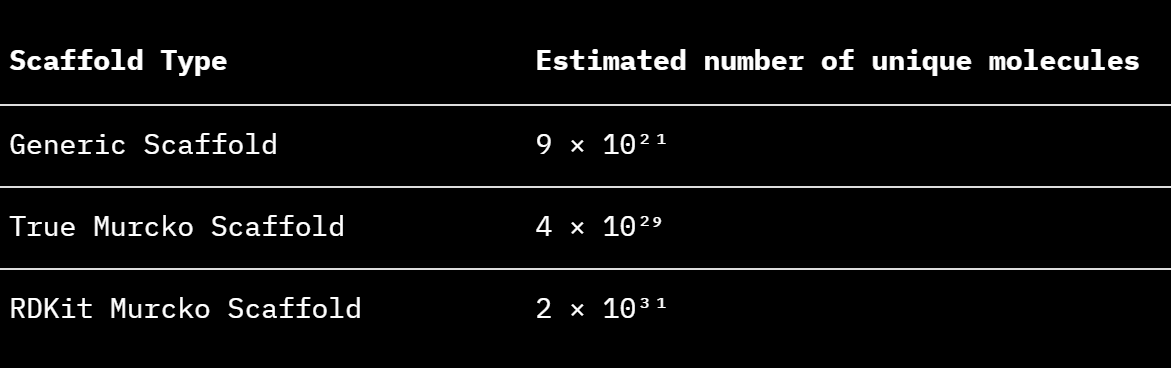
Molecular Diversity Average Tanimoto dissimilarity via ECFP4 fingerprints is strikingly high—0.889 for full molecules, 0.871 for RDKit Murcko scaffolds, 0.869 for True Murcko scaffolds, and 0.626 for Generic scaffolds. The uniqueness depends on the sample size, and with larger samples there is a greater risk of generating duplicates which lowers the overall uniqueness. Nonetheless, the uniqueness of 1 billion samples remains high: 75.3% of full molecules, 27.3% of RDKit Murcko scaffolds, 24.4% of True Murcko scaffolds, and 3.32% of Generic scaffolds.
Conclusion
This analysis concludes that the explorable chemical space of Anyo lab’s MolGen estimated to be as large as 10²⁶ (with a 95% confidence interval between 10²³ and 10²⁹), with its molecules and scaffolds exhibiting exceptional diversity. Yet, the methods' inability to pinpoint the true size—potentially magnitudes larger—highlights the tool's innovative depth. Adjusting the sampling temperature parameter could amplify the uniqueness by favoring low-probability gems, a tunable innovation that elevates Molecule Generator above scaffold-dependent models.
In practice, MolGen's size and diversity enable screening of vast and diverse molecules, allowing researchers to apply multiple filters—such as synthetic accessibility, ADMET properties, and novelty—more effectively, thereby identifying lead-like hits with greater efficiency and precision. This expansive scale has proven instrumental in real-life applications, including analogue generation for lead optimization requiring innovative scaffolds, as well as massive de novo lead-like hit identification. The diversity not only fosters novel molecule discovery through unique scaffolds but also ensures experimentally validated high synthesizability, accelerating the transition from virtual screening to laboratory validation.
References
[1] Bohacek, R. S., McMartin, C., & Guida, W. C. (1996). The art and practice of structure‐ based drug design: a molecular modeling perspective. Medicinal research reviews, 16(1), 3- 50.
[2] Chao, A. (1984). Nonparametric estimation of the number of classes in a population. Scandinavian Journal of statistics, 265-270.
[3] Chao, A., & Lee, S. M. (1992). Estimating the number of classes via sample coverage. Journal of the American statistical Association, 87(417), 210-217.
[4] Good, I. J. (1953). The population frequencies of species and the estimation of population parameters. Biometrika, 40(3-4), 237-264.